









How do Mitochondia Replicate?
The Mitochondrial replication is diagrammed in the cartoon in the side bar and shown above in an electron micrograph. Mitochondria replicate much like bacterial cells. When they get too large, they undergo fission. This involves a furrowing of the inner and then the outer membrane as if someone was pinching the mitochondrion. Then the two daughter mitochondria split. Of course, the mitochondria must first replicate their DNA. This will be discussed in more detail in the next section. An electron micrograph depicting the furrowing process is shown in these figures. The figure above was taken from Fawcett, A Textbook of Histology, Chapman and Hall, 12th edition, 1994

Sometimes new mitochondria are synthesized de novo in centers that are rich in proteins and polyribosomes needed for their synthesis. The electron micrograph in the above figure shows such a center. It appears that the cluster of mitochondria are sitting in a matrix of proteins and other materials needed for their production. How might you prove that material in that region was making mitochondrial proteins? Return to Menu
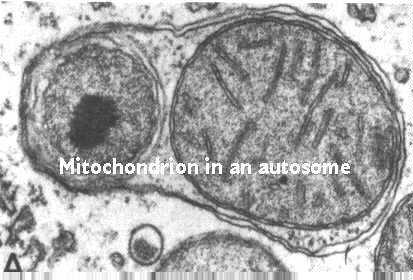
Certain mitochondrial proteins are needed before the mitochondria can divide.
This has been shown in a study by Sorgo and Yaffe, J Cell Bio. 126: 1361-1373, 1994. They showed the result of the removal of an outer membrane protein from mitochondria called MDM10. This figure shows the results. The mitochondria are able to take in components and produce membranes and matrix enzymes. However, fission is not allowed. Thus, the result is a giant mitochondrion. This is illstrated in the micrograph below.

Mitochondrial DNA and its function.
Mitochondria have some of their own DNA, ribosomes, and can make many of their own proteins. The DNA is circular and lies in the matrix.in punctate structures called "nucleoids". Each nucleoid may contain 4-5 copies of the mitochondrial DNA (mrDNA).
Human mitochondrial DNA is 16,569 bp; encodes a number of mitochondrial proteins
- Subunits 1, 2, and 3 of cytochrome oxidase
- Subunits 6, 8,9 of the Fo ATPase
- Apocytochrome b subunit of CoQH2-Cytochrome C reductase
- Seven NADH-CoQ reductase subunits
The nucleus encodes the remaining proteins. Most of the lipid is imported (recall the lectures on lipid addition to membranes). This cartoon from your text shows the nuclear involvement. The highlighted labels are drugs that can be used to block the process and test the source of the mitochondrial protein.

Mitochondria also have their own ribosomes and tRNA:
- 22 tRNAs
- rRNAs
- 16S
- 12S
- 5S
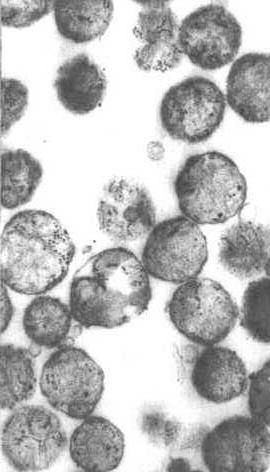
(Magalhaes, PJ; Andreu, AL, Schon EA, Evidence for the presence of 5 S rRNA in mammalian mitochondria Mol Biol Cell 9: 2375-2382)
The Figure to the left shows mitochondrial Ribosomes as granules in the mitochondria.
The texts still say that mitochondria have no 5S rRNA, however the recent study cited above shows evidence for 5S in carefully prepared mitochondrial fractions. These workers found 5S in highly purified mitochondria and mitoplasts (mitochondria without the outer membrane). Conclusion: 5S rRNA is imported into mitochondria, but its function is uncertain.
Visualization of mitochondrial DNA
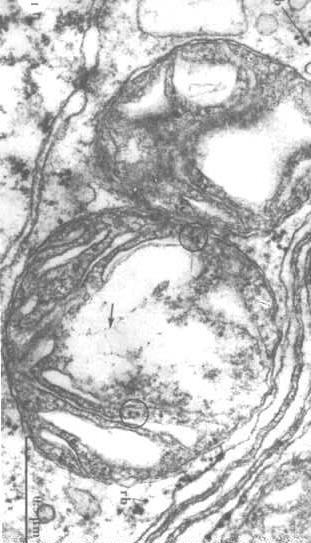
To
visualize the structure of mitochondrial DNA, we have to extract the proteins in the matrix and reveal the DNA (arrows in the figure to the right).
structure of mitochondrial DNA, we have to extract the proteins in the matrix and reveal the DNA (arrows in the figure to the right).
One can also see ribosomes in the circles.
Alternatively, one can extract the DNA and float it on a water surface. Then, it can be picked up by a plastic coated grid, and examined in the electron microscope. Mitochondrial circular DNA is shown in the following figure. This electron micrograph is taken from Fawcett, A Textbook of Histology, Chapman and Hall, 12th edition, 1994.
Mitochondrial Inheritance
In mammals, 99.99% of mitochondrial DNA (mtDNA) is inherited from the mother. This is because the sperm carries its mitochondria around a portion of its tail and has only about 100 mitochondria compared to 100,000 in the oocyte. As the cells develop, more and more of the mtDNA from males is diluted out. Hence less than one part in 104 or 0.01% of the mtDNA is paternal. This means that mutations of mtDNA can be passed from mother to child. It also has implications if one does cloning of mammals with the use of somatic cells. The nuclear DNA would be from the donor cell, but the mtDNA would be from the host cell. This is how Dolly the sheep was cloned.
There is a Yeast strain, called "Petite" that have structurally abnormal mitochondria that are incapable of oxidative phosphorylation. These mitochondria have lost some or all of their DNA. Mitochondrial inheritance from yeast is biparental, and both parent cells contribute to the daughter cells when the haploid cells fuse. After meiosis and mitosis, there is random distribution of mitochondria to daughter cells. If the fusion is with yeast that are petite and yeast that are not, a certain percentage of the daughter cells will be "petite".
Mutations in mammalian mtDNA do cause diseases, because there is such a short sequence and very heavy information content in the sequence. The next lecturer on mitochondria in this series will spend a great deal of time on the mitochondrial genome. Since each cell contains hundreds of mitochondria and thousands of copies of the genome, the effects of the mutated mitochondria may be diluted out. As expected, those tissues or organs most likely to be affected would be the ones most dependent on oxidative phosphorylation (ATP production). In young persons it might not be picked up because even a person with 15% normal mitochondria might have enough to be healthy. However, aging patients may show a more severe disease phenotype.
Some example of diseases:
- Leber's hereditary optic neuropathy (degeneration of the optic nerve, accompanied by increasing blindness): caused by mutation to the gene encoding subunit 4 of the NADH-C0Q reductase.
- "ragged muscle fibers" associated with jerky movements is caused by mutation of mitochondrial lysine tRNA.
- Kaerns-Sayre syndrome: eye defects, abnormal heartbeat, Central nervous system degeneration. Several large deletions in mtDNA.
Can damaged mitochondrial DNA be repaired?
Current studies say yes.
- Meeusen, S, Tieu, Q, Wong E, Weiss, E, Schieltz, D, Yates, JR, and Nunnari, J. Mgm101p is a novel component of the mitochonrial nucleoid that binds DNA and is required for the repair of oxidatively damaged mitochondrial DNA. J Cell Biol 145: 291-304 (1999)
- Mgm stands for "mitochondrial genome maintenance". It was discovered in yeast cells while searching for mutants that caused a temperature sensitive loss of mitochondrial DNA.
- Fused Mgm101 to green fluorescent protein and found that it was localized to the punctate "nucleoid" structures. Localization overlapped with that of DNA detection systems.
- After protein screening found the Mgm101, they studied how its loss affected respiratory competence. Clearly the protein was needed for function, but they do not know exactly what its role is at this point.
- Looked at the COOH terminal region and saw that it was highly basic. That suggested that the Mgm101p might have the ability to bind DNA. Compared its binding to DNA cellulose columns (in high salt conditions) with another known DNA binding protein and confirmed relatively high affinity binding by both proteins.
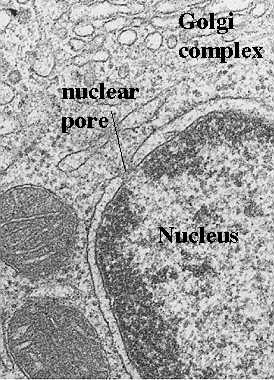
What happens to old, worn-out mitochondria?
Mitochondrial numbers are controlled by autophagy. This is a process by which lysosomes are involved in controlling cell constituents. This Figure shows the process; it is taken from Fawcett, A Textbook of Histology, Chapman and Hall, 12th edition, 1994.
The process begins by wrapping endoplasmic reticulum membranes around the mitochondrion. Then, vesicles come from the Golgi complex and join with the autophagic vacuole. These vesicles contain hydrolases attached to the mannose 6 phosphate receptors in the vesicle membranes. The lysosome web page discusses their function and fate. Recall that they fuse with the autophagic vacuole. The acid pH then allows the hydrolases to be removed from their receptors. The receptors are recycled back to the Golgi complex in other vesicles.
In the meantime, the lysosome forms as the pH drops and the cells begin to degrade the contents. Recall that lysosomes are LAMP+, but they do not carry the MPR because these have been recycled to the Golgi Complex. What coat is found around the transport vesicles going to the autophagic vacuole?
For more information, contact:
Gwen Childs, Ph.D.,FAAA
Professor and Chair
Department of Neurobiology and Developmental Sciences
University of Arkansas for Medical Sciences
Little Rock, AR 72205
For questions, contact this email address:
